
Medikamente bei pulmonal-arterieller Hypertonie
- Autor(en): Urspeter Masche
- pharma-kritik-Jahrgang 39
, Nummer 6, PK1025
Redaktionsschluss: 5. Oktober 2017
DOI: https://doi.org/10.37667/pk.2017.1025 - PDF-Download der Printversion dieser pharma-kritik Nummer
Pulmonale Hypertonien – definiert durch eine Erhöhung des pulmonal-arteriellen Mitteldrucks auf mindestens 25 mm Hg – werden pathophysiologisch und klinisch in fünf Gruppen unterteilt: Gruppe 1 bilden die pulmonal-arteriellen Hypertonien (mit denen sich diese Übersicht im Weiteren befasst), Gruppe 2 die pulmonalen Hypertonien bei Linksherzerkrankungen, Gruppe 3 die pulmonalen Hypertonien bei Lungenerkrankungen, Gruppe 4 die pulmonalen Hypertonien bei chronischer Thromboembolie und Gruppe 5 die pulmonalen Hypertonien, denen unklare oder verschiedenartige Mechanismen zugrundeliegen (z.B. bei hämatologischen Erkrankungen oder Sarkoidose).
Pulmonal-arterielle Hypertonien, deren Häufigkeit auf ungefähr 1,5 pro 100’000 geschätzt wird, sind charakterisiert durch eine endotheliale Dysfunktion mit einer Dysbalance von vasoaktiven Substanzen (vermehrte Bildung von Endothelin-1, verminderte Synthese von Stickstoffmonoxid und Prostazyklin). Es entwickelt sich eine Verengung der kleinen Pulmonalarterien, die mit einer Proliferation und Fibrose der Intima, Hypertrophie der Media, perivaskulären Infiltraten und Thrombenbildung einhergeht. Die Zunahme des Gefässwiderstands kann mit der Zeit zur Rechtsherzüberlastung führen. Ätiologisch werden bei der pulmonal-arteriellen Hypertonie vier Formen unterschieden: (1) die idiopathische Form; (2) die hereditäre Form (zu vermuten, wenn in einer Familie mehrere Fälle auftreten); (3) die medikamenten- oder toxinbedingte Form (z.B. durch Appetitzügler); (4) Formen, die mit anderen Krankheiten assoziiert sind, wie Kollagenosen, HIV-Erkrankung, portaler Hypertonie und kongenitalen Herzvitien.
Das Hauptsymtpom der pulmonal-arteriellen Hypertonie ist die Dyspnoe unter Belastung; in fortgeschrittenen Stadien kommen Müdigkeit, Synkopen, Brustschmerzen und Beinödeme hinzu. Da die Symptome wenig spezifisch sind, bedarf es einer niedrigen Verdachtsschwelle, um an die Diagnose zu denken. Erste und in der hausärztlichen Praxis durchführbare Untersuchungen sind ein EKG, Thorax-Röntgenbild, Lungenfunktionstest und die Bestimmung von BNP oder NT-proBNP. Die Kombination von EKG und BNP scheint bereits eine gute Sensitivität zu besitzen. Wichtig ist die Echokardiographie, mit der sich die Druckverhältnisse im kleinen Kreislauf abschätzen lassen. Die Diagnose wird mit der Rechtsherzkatheter-Untersuchung gestellt, die in einem spezialisierten Zentrum erfolgen sollte; sie ist vor Therapiebeginn obligat, unter anderem weil damit zum Beispiel eine postkapilläre pulmonale Hypertonie ausgeschlossen wird. Weitere Funktionstests, Laboranalysen und radiologische Untersuchungen helfen zur Differenzierung der pulmonal-arteriellen Hypertonie. Elemente, die zur prognostischen Beurteilung und auch zur Verlaufs- oder Therapiekontrolle dienen, sind der funktionelle Schweregrad (der analog zur NYHA-Einteilung in vier Stadien unterteilt wird) und der 6-Minuten-Gehtest.
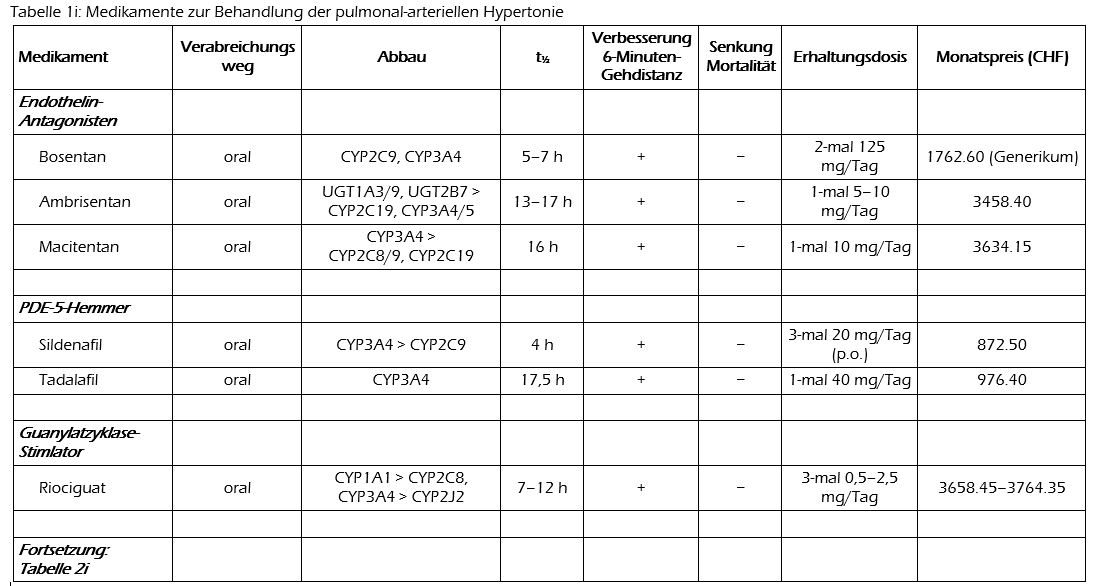
Die Behandlung einer pulmonal-arteriellen Hypertonie setzt sich zusammen aus allgemeinen und aus medikamentösen Mass-nahmen. Bei den Medikamenten unterscheidet man unspezifisch wirkende von solchen, die direkt in die Mechanismen eingreifen, welche die Vasokonstriktion verursachen. (Diese spezifisch wirkenden Medikamente werden weiter unten näher beschrieben, zusätzliche Informationen liefert die im Internet publizierte Tabelle 1i.) Um die Wirkung der Medikamente zu erfassen, eignet sich im klinischen Alltag der 6-Minuten-Gehtest, der auch in der Mehrheit der klinischen Studien als primärer Endpunkt festgelegt wurde. Was als klinisch relevante Verbesserung der 6-Minuten-Gehstrecke gilt, ist allerdings nicht exakt bestimmt. Um den Nutzen der Medikamente im Langzeitverlauf zu dokumentieren, werden auch zunehmend «härtere» Endpunkte wie die Kombination von Hospitalisationsrate und Mortalität berücksichtigt.
Therapieziel ist es, den Patienten oder die Patientin in die sogenannte Niedrigrisikogruppe zu überführen, bei der das 1-Jahres-Sterberisiko unter 5% liegt.(1) Die Zuteilung zur Niedrigrisikogruppe wird unter anderem dadurch bestimmt, dass keine oder höchstens minimale Zeichen einer Rechtsherzinsuffizienz bestehen, keine Synkopen auftreten, ein NYHA-Stadium I oder II vorliegt und die 6-Minuten-Gehstrecke mehr als 440 m beträgt – wobei es natürlich mit fortschreitender Erkrankung schwieriger wird, diese Ziele zu erreichen.
Allgemeinmassnahmen
Schon allein, weil es sich bei der pulmonal-arteriellen Hypertonie um eine seltene Krankheit handelt, die eine komplexe Behandlung erfordert, sollte die Betreuung der betroffenen Patienten und Patientinnen unter Führung eines spezialisierten Zentrums stattfinden.
Während früher aus Furcht vor Synkopen und Rechtsherzversagen von körperlicher Belastung abgeraten wurde, weiss man heute, dass ein adaptiertes Training zur Verbesserung von Leistungsfähigkeit und Lebensqualität beiträgt.(2) Hierfür empfehlen sich Fachkliniken, die im Rahmen einer pulmonalen Rehabilitation ein strukturiertes Programm anbieten.
Bei der Sauerstoffsättigung sollte man einen Wert von mindestens 90% anstreben. Bei Leuten mit einer pulmonal-arteriellen Hypertonie liess sich durch Sauerstoffverabreichung die Belastbarkeit bei der Ergometrie verbessern.(3) Über den allfälligen Nutzen einer längerfristigen Sauerstoffbehandlung sind aber noch keine Daten publiziert.
Eine Salzrestriktion kann einer Volumenüberlastung vorbeugen, die im Zusammenhang mit einer Rechtsherzinsuffizienz auftreten kann. Ferner ist auf die empfohlenen Impfungen zu achten (Influenza, Pneumokokken). Eine Schwangerschaft bedeutet für eine Frau mit pulmonal-arterieller Hypertonie ein hohes Risiko und ist unbedingt zu vermeiden.
Wichtig ist auch die psychosoziale Unterstützung, die man den Patienten und Patientinnen anbieten soll.
Unspezifische medikamentöse Massnahmen
Schleifendiuretika helfen, das intravasale Volumen zu vermindern und den rechten Ventrikel zu entlasten. Häufig werden auch Aldosteronantagonisten eingesetzt. Digoxin kann die kardiale Funktion verbessern, wird aber nur noch ausnahmsweise verwendet. Die Anwendung dieser Substanzen ist nicht durch kontrollierte Studien überprüft.
Bei der pulmonal-arteriellen Hypertonie treten intrapulmonale (Mikro-)Thrombosen auf, was Antikoagulantien ins Spiel bringt. Ihre Anwendung ist allerdings nur in Beobachtungsstudien untersucht worden. Für die idiopathische Form der pulmonal-arteriellen Hypertonie sind die Ergebnisse widersprüchlich; es gibt sowohl Daten, die unter einer Antikoagulation einen Überlebensvorteil beschreiben, als auch solche, bei denen dies nicht der Fall ist. Bei pulmonal-arteriellen Hypertonien, die durch eine Bindegewebserkrankung bedingt sind, scheint die Antikoagulation die Überlebenswahrscheinlichkeit sogar zu vermindern.(4,5) Eine Antikoagulation wird deshalb – bei mittelmässiger Evidenzlage – nur für die idiopathische, hereditäre und medikamenteninduzierte Form der pulmonal-arteriellen Hypertonie empfohlen.
Etwa 5 bis 10% der Personen mit einer idiopathischen pulmonal-arteriellen Hypertonie reagieren positiv auf einen dynamischen Test mit einer vasodilatierenden Substanz (z.B. inhaliertes Stickstoffmonoxid), den man anlässlich der Rechtsherzkatheter-Untersuchung durchführt. Solche «vasoreaktiven« Fälle sollten primär mit Kalziumantagonisten behandelt werden, und zwar solange, als sie sich im NYHA-Stadium I oder II befinden.
Beeinflussung des Endothelin-Signalwegs
Endotheline sind Peptidhormone, deren Wirkung über die beiden Endothelinrezeptor-Typen A und B vermittelt wird. Endothelin-1, das bei pulmonal-arterieller Hypertonie vermehrt gebildet wird, ist ein starker Vasokonstriktor und fördert auch die Zellproliferation.
Bosentan (Tracleer® u.a.) ist der Prototyp der oral verabreichbaren Endothelinrezeptor-Antagonisten und blockiert sowohl den Endothelin-A- wie den -B-Rezeptor. Es ist ein Zytochrom-Induktor, der den eigenen Abbau beschleunigt. Wie eine Metaanalyse zeigte, verbessert Bosentan den pulmonal-arteriellen Mitteldruck und die 6-Minuten-Gehstrecke signifikant; die Differenz zu Placebo betrug 6 mm Hg bzw. 46 m. Auch der allgemeine klinische Zustand wurde durch Bosentan günstig beeinflusst. Keinen Unterschied stellte man bei der Gesamtmortalität fest.(6)
Macitentan (Opsumit®) ist ebenfalls ein Endothelin-A/B-Antagonist. In einer Dosis von 3 oder 10 mg/Tag wurde es in einer grossen Doppelblindstudie mit Placebo verglichen. Die durchschnittliche Behandlungsdauer betrug 2,2 Jahre (Medianwert). Fälle einer relevanten Krankheitsverschlechterung sowie Todesfälle bildeten in Kombination den primären Studienendpunkt. Die Häufigkeit dieser Ereignisse betrug in den Macitentan-Gruppen 38% bzw. 31% und in der Placebo-Gruppe 46%.(7)
Ambrisentan (Volibris®), ein selektiver Endothelin-A-Antagonist, wurde in zwei placebokontrollierten Doppelblindstudien untersucht, die 12 Wochen dauerten und gemeinsam publiziert wurden. In beiden Studien wurde der primäre Endpunkt, die 6-Minuten-Gehstrecke, durch Ambrisentan signifikant besser beeinflusst als durch Placebo.(8)
Als Nebenwirkungen von Endothelin-Antagonisten sind Kopfschmerzen, Ödeme, Hitzewallungen und Hautrötung, Schwindel, Übelkeit, Stuhlunregelmässigkeiten, Anämie, Synkopen, Nasopharyngitis und Leberenzymerhöhungen beschrieben. Insbesondere bei Bosentan sind auch manifeste Leberschädigungen vorgekommen, so dass die Behandlung von regelmässigen Leberwertkontrollen begleitet sein sollte. Endothelin-Antagonisten sind teratogen.
Beeinflussung des Stickstoffmonoxid-Signalwegs
Phosphodiesterase-Hemmer
In Endothelzellen und in Epithelien der Atemwege wird aus der Aminosäure Arginin Stickstoffmonoxid gebildet, das die Umwandlung von Guanosin-Triphosphat (GTP) in das gefässerweiternd und antiproliferativ wirkende zyklische Guanosin-Monophosphat (cGMP) katalysiert. Die Inaktivierung von cGMP findet durch Phosphodiesterasen statt, wobei in den glatten Gefässmuskelzellen der Typ 5 dominiert.
Selektive Hemmer der Phosphodiesterase-5 (PDE-5) sind ursprünglich zur Behandlung der erektilen Dysfunktion eingeführt worden. Unterdessen sind sie zum Teil auch für die Behandlung der pulmonal-arteriellen Hypertonie zugelassen.
Sildenafil wird unter dem Namen Revatio® zur oralen und intravenösen Therapie der pulmonal-arteriellen Hypertonie angeboten (die Injektionslösung ist in der Schweiz nicht kassenpflichtig). In einer Metaanalyse wurden vier placebokontrollierte Doppelblindstudien berücksichtigt, in denen Sildenafil (3-mal 20–80 mg/Tag) während 12 bis 16 Wochen bei pulmonal-arterieller Hypertonie eingesetzt worden war. Dabei wurde nachgewiesen, dass Sildenafil die 6-Minuten-Gehstrecke um durchschnittlich 31 m verbessert und das Risiko einer Zustandsverschlechterung signifikant senkt. Bei der Mortalität wurde kein Unterschied gegenüber Placebo festgestellt.(9)
Tadalafil ist als Adcirca® zur oralen Behandlung der pulmonal-arteriellen Hypertonie erhältlich. In einer placebokontrollierten Doppelblindstudie, die 16 Wochen dauerte, zeigte sich, dass auch Tadalafil dosisabhängig die 6-Minuten-Gehstrecke zu verbessern vermag.(10)
Die häufigsten Nebenwirkungen von PDE-5-Hemmern sind Kopfschmerzen, Hitzewallungen und Hautrötung, Muskelschmerzen, verstopfte Nase, Dyspepsie, Blutdrucksenkung sowie Sehstörungen. Möglicherweise können PDE-5-Hemmer zu einer Erhöhung des Melanom-Risikos beitragen.(11)Wegen der Gefahr von schweren Hypotonien dürfen PDE-5-Hemmer nicht mit Nitraten oder Molsidomin (Corvaton®) kombiniert werden.
Guanylatzyklase-Stimulatoren
Das oral verabreichbare Riociguat (Adempas®) stimuliert die Guanylatzyklase, die GTP zu cGMP umwandelt. Die Hauptstudie zur Anwendung bei pulmonal-arterieller Hypertonie umfasste 443 Personen, die man mit Riociguat (3-mal täglich 0,5–2,5 mg) oder Placebo behandelte. Nach 12 Wochen hatte die 6-Minuten-Gehstrecke mit Riociguat um 30 m zu- und mit Placebo um 6 m abgenommen.(12)
Als Nebenwirkungen sind Kopfschmerzen, Dyspepsie, Schwindel, Übelkeit und Erbrechen, Durchfall, periphere Ödeme, Hypotonie und Synkopen zu nennen. Ferner sind Einzelfälle einer Hämoptyse aufgetreten.
Riociguat soll wegen der erhöhten Gefahr einer Hypotonie nicht mit PDE-5-Hemmern kombiniert werden. Bei Leuten, die rauchen, ist wegen der CYP1A1-Induktion mit einer verminderten Riociguat-Wirkung zu rechnen.
Beeinflussung des Prostazyklin-Signalwegs
Prostazyklin-Analoga
Prostazyklin (Prostaglandin I2) wirkt, nachdem es sich an den Prostazyklin-Rezeptor gebunden und eine Signalkaskade in Gang gesetzt hat, gefässerweiternd, antithrombotisch, antiproliferativ und entzündungshemmend. Bei pulmonal-arterieller Hypertonie ist wegen einer verminderten Expression der Prostazyklin-Synthase die Produktion von Prostazyklin vermindert.
Prostazyklin-Analoga bilden seit Langem einen wichtigen Baustein bei der Behandlung der pulmonal-arteriellen Hypertonie. Es sind relativ instabile Substanzen mit einer kurzen Halbwertszeit, weshalb sie nicht oral verabreicht werden können und gute Kenntnisse bei der Verabreichung und Dosierung erfordern.
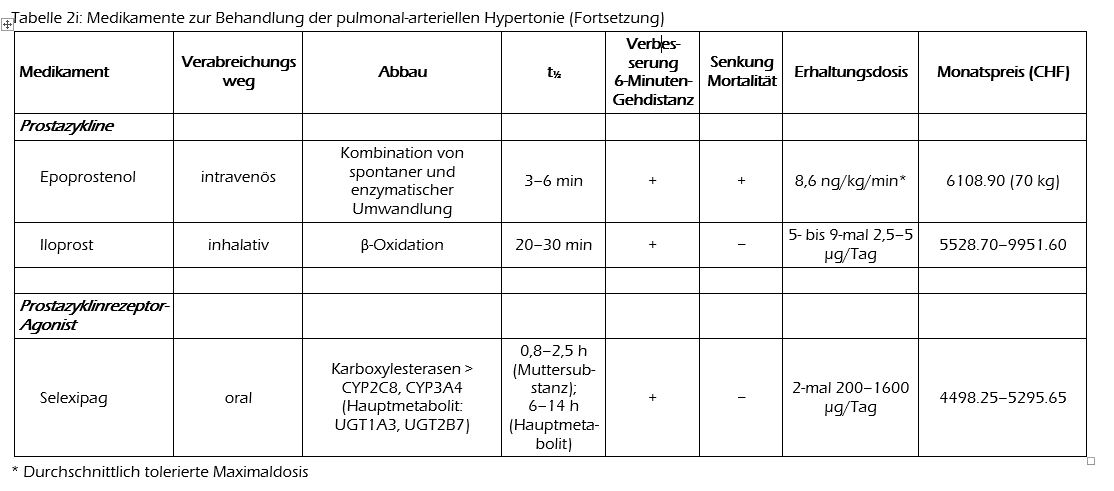
Bei Epoprostenol (Flolan®, Veletri®) handelt es sich um synthetisch hergestelltes Prostazyklin. Es kann nur über einen zentralvenösen Katheter als Dauerinfusion verabreicht werden. Zu Epoprostenol liegen zwei randomisierte Studien vor, in denen das Medikament während 12 Wochen verabreicht wurde. In der einen Studie (n=81) wurden Personen mit einer idiopathischen pulmonal-arteriellen Hypertonie behandelt,(13) in der zweiten solche mit einer durch eine Sklerodermie bedingten pulmonal-arteriellen Hypertonie (n=111).(14) In beiden Studien liess sich die 6-Minuten-Gehstrecke mit Epoprostenol signifikant verbessern. In der erstgenannten Studie wurde zudem auch ein Unterschied bei der Mortalität festgestellt. Angesichts dessen, dass diese Studie nur 81 Individuen umfasste und 12 Wochen dauerte, muss man eine Aussage zur Mortalitätsreduktion aber vorsichtig bewerten.
Iloprost (Ventavis®) ist ein Prostazyklin-Analogon, das chemisch stabiler ist als Epoprostenol. Es wird via Inhalation angewendet (mit Hilfe von Verneblern). Die Hauptstudie, die mit Iloprost durchgeführt wurde, umfasste 203 Personen und ergab nach 12 Wochen einen signifikanten Unterschied bei der 6-Minuten-Gehstrecke im Vergleich zu Placebo.(15)
Auch Treprostinil (Remodulin®) ist ein Prostazyklin-Analogon und steht zur subkutanen oder intravenösen Verabreichung zur Verfügung. Die subkutane Gabe von Treprostinil wurde in einer placebokontrollierten Doppelblindstudie geprüft, wobei sich nach 12 Wochen eine signifikante Verbesserung der 6-Minuten-Gehstrecke erzielen liess.(16)
Zu den Nebenwirkungen von Prostazyklin-Analoga gehören Kopfschmerzen, Kieferschmerzen, Übelkeit, Erbrechen, Durchfall, Vasodilatation, Hypotonie, Schwindel, Hautausschläge, Muskel- und Gelenkschmerzen, Brustschmerzen, Angstzustände, Brady- oder Tachykardie, Husten sowie periphere Ödeme. Bei Treprostinil ist Vorsicht angezeigt, wenn es zusammen mit CYP2C8-Hemmern verabreicht wird.
Prostazyklinrezeptor-Agonisten
Selexipag (Uptravi®) unterscheidet sich chemisch von den Prostazyklin-Analoga, wirkt aber ebenfalls als Agonist am Prostazyklin-Rezeptor. Es wird oral verabreicht und in der Leber durch Karboxylesterasen zum Metaboliten ACT-333679 (MRE-269) hydrolysiert, der um ein Vielfaches potenter als die Muttersubstanz und somit für die Hauptwirkung verantwortlich ist. Die Zulassung von Selexipag beruht auf einer Doppelblindstudie, in der 1156 Personen Selexipag (max. 2-mal 1600 mcg/Tag) oder ein Placebo erhielten. Primärer Endpunkt war die Kombination aus relevanter Krankheitsverschlechterung und Tod; die 6-Minuten-Gehstrecke wurde als sekundärer Endpunkt gewertet. Die durchschnittliche Beobachtungszeit betrug 71 Wochen in der Selexipag- bzw. 64 Wochen in der Placebo-Gruppe (Medianwerte). Ein zum primären Endpunkt zählendes Ereignis trat unter Selexipag bei 27% der Behandelten auf, unter Placebo bei 42%, was einer «Hazard Ratio» von 0,60 (99% CI 0,46–0,78) entsprach. Nahm man die Gesamtmortalität allein, unterschieden sich die beiden Gruppen aber nicht signifikant (18,0 gegenüber 17,4%). Bei der 6-Minuten-Gehstrecke ergab sich ein Unterschied von 12 m zugunsten von Selexipag.(17)
Bei Selexipag treten ähnliche Nebenwirkungen auf wie bei Prostazyklin-Analoga; ausserdem sind unter Selexipag Fälle von Hyperthyreose beobachtet worden. Auch bei Selexipag ist auf mögliche Interaktionen mit CYP2C8-Hemmern zu achten; die Kombination mit Gemfibrozil (Gevilon®), einem starken CYP2C8-Hemmer, ist kontraindiziert.
Kombinationsbehandlung
Analog zu anderen Erkrankungen lässt bei der pulmonal-arteriellen Hypertonie die Kombination von verschiedenen Substanzen synergistische Effekte erhoffen. Einige der oben zitierten Studien entsprachen im Prinzip einer Kombinationsbehandlung, weil die Prüfsubstanzen mehrheitlich als Zusatz zu einer bestehenden Behandlung mit spezifisch wirkenden Medikamenten verordnet wurden. Es existieren aber auch einige grössere randomisierte Studien, die sich gezielt der Frage der Kombinationsbehandlung widmeten.
In drei placebokontrollierten Doppelblindstudien, 12 bis 16 Wochen dauernd, erwies sich eine Kombinationsbehandlung gegenüber einer Monotherapie beim 6-Minuten-Gehtest als signifikant überlegen: In einer Studie wurde einer Behandlung mit Sildenafil oder Bosentan Treprostinil (als Inhalation) hinzugefügt.(18) In einer anderen Studie wurde Epoprostenol (intravenös) mit hochdosiertem Sildenafil kombiniert.(19) In der dritten Studie wurde die zusätzliche Verabreichung von Tadalafil bei Personen unter Bosentan untersucht.(20)
In zwei anderen Doppelblindstudien liessen sich demgegenüber keine signifikanten Verbesserungen dokumentieren, weder bei der 6-Minuten-Gehstrecke (bei Kranken unter einem Endothelin-Antagonisten oder einem PDE-5-Hemmer, bei denen man die zusätzliche Gabe von oralem Treprostinil testete),(21) noch beim Krankheitsverlauf (bei Leuten unter Sildenafil, die zusätzlich 3 Jahre lang Bosentan verwendet hatten).(22)
Alle diese Studien befassten sich mit Individuen, die bereits unter einer Behandlung (Monotherapie) standen und bei denen wegen ungenügender Wirkung oder Zustandsverschlechterung eine Therapieausweitung indiziert war. Was der Nutzen ist, wenn man gleich zu Beginn kombiniert behandelt, wurde in einer grossen Studie untersucht. 500 Personen (31% NYHA II, 69% NYHA III), die noch keine spezifische Therapie erhalten hatten, wurden doppelblind auf drei Gruppen verteilt: die erste Gruppe erhielt Ambrisentan (10 mg/Tag) plus Tadalafil (40 mg täglich), die zweite Ambrisentan allein und die dritte Tadalafil allein. Als primärer Endpunkt zählten Tod, relevante Krankheitsverschlechterung oder ungenügendes klinisches Ansprechen. Nach einer durchschnittlichen Beobachtungszeit von ungefähr 20 Monaten war in der Ambrisentan/Tadalafil-Gruppe bei 18% eines dieser Ereignisse eingetreten (Zahl der Todesfälle 4%), in der Ambrisentan-Gruppe bei 34% (2%) und in der Tadalafil-Gruppe bei 28% (5%).(23)
Behandlungsrichtlinien
Bei allen Patienten und Patientinnen sollten nach der Diagnosestellung unverzüglich die allgemeinen oder unspezifischen Massnahmen eingeleitet werden. Wenn die Krankheit das NYHA-Stadium II erreicht hat, sollte man zu einem der spezifisch wirkenden Medikamente greifen. Welche dieser Substanzen sich für die initiale Behandlung am besten eignet, ist nicht bestimmt, da weder Direktvergleiche vorliegen noch persönliche Merkmale identifiziert sind, die das individuelle Ansprechen vorhersehen lassen. Den Ausschlag geben neben praktischen Überlegungen die Erfahrungen des Behandlungsteams und die individuelle Verträglichkeit. Im Stadium II ist eine orale Behandlung zu bevorzugen, während die inhalative oder intravenöse Behandlung für die Stadien III und IV reserviert bleibt (wobei die inhalative Behandlung an Bedeutung verloren hat).
Alle drei Monate muss überprüft werden, ob das Behandlungsziel erreicht worden ist. Falls nicht, sollte ein zusätzliches Medikament aus einer anderen Stoffgruppe verordnet werden. Welcher Stellenwert der initialen Kombinationstherapie zukommt, ist noch nicht definiert; besonders für Individuen, die sich bei der Diagnose bereits in einem fortgeschritteneren Stadium befinden, könnte es sich um eine vorteilhafte Modalität handeln. Viele Fachleute propagieren, dass man sich wegen der generell schlechten Prognose der pulmonal-arteriellen Hypertonie rasch auf eine Kombinationsbehandlung stützen soll.
Wenn auch eine ausgebaute medikamentöse Therapie nicht genügend hilft oder sich Symptome rasch verschlechtern, kann sich eine Lungentransplantation anbieten, sofern die Altersgrenze von 60 Jahren nicht überschritten und die Lunge als einziges wichtiges Organ erkrankt ist. Eine weitere Option – meist als Überbrückung bis zur Transplantation – ist die atriale Septostomie, ein perkutan durchgeführter Eingriff, mit dem ein Rechts-links-Shunt geschaffen und die Hämodynamik verbessert wird.
Pulmonal-arterielle Hypertonien kommen auch bei Kindern vor. Weil für die Behandlung bei Kindern praktisch keine Studiendaten vorhanden sind, gelten mehr oder weniger die gleichen Richtlinien wie für Erwachsene.
Schlussfolgerungen
Medikamente, mit denen man bei pulmonal-arterieller Hypertonie in die pathophysiologischen Mechanismen eingreift, können Symptome lindern, die Leistungsfähigkeit steigern und den Krankheitsverlauf verlangsamen. Mit der Behandlung visiert man eine Krankheitsstufe an, die funktionell einem Normalzustand möglichst nahekommt. An diesem Ziel orientiert sich die Intensität der Behandlung, das heisst, dass allenfalls auch verschiedene Medikamente miteinander zu kombinieren sind.
Obschon sich die medikamentösen Möglichkeiten bei der pulmonal-arteriellen Hypertonie erweitert haben, bleibt es in den meisten Fällen eine fortschreitende und mit schlechter Prognose behaftete Erkrankung. Zwar wird von Fachleuten aufgrund ihrer Erfahrungen bejaht, dass mit einer heutigen Behandlung auch die Mortalität gesenkt wird. Diese Beobachtung ist aber mit prospektiven Daten relativ wenig abgesichert.
Literatur
- 1) Galiè N et al. Eur Heart J 2016; 37: 67-119
- 2) Pandey A et al. Circ Heart Fail 2015; 8: 1032-43
- 3) Ulrich S et al. Eur Heart J 2017; 38: 1159-68
- 4) Olsson KM et al. Circulation 2014; 129: 57-65
- 5) Preston IR et al. Circulation 2015; 132: 2403-11
- 6) Lee YH, Song GG. Korean J Intern Med 2013; 28: 701-7
- 7) Pulido T et al. N Engl J Med 2013; 369: 809-18
- 8) Galiè N et al. Circulation 2008; 117: 3010-9
- 9) Wang RC et al. Respir Med 2014; 108: 531-7
- 10) Galiè N et al. Circulation 2009; 119: 2894-903
- 11) Li WQ et al. JAMA Intern Med 2014; 174: 964-70
- 12) Ghofrani HA et al. N Engl J Med 2013; 369: 330-40
- 13) Barst RJ et al. N Engl J Med 1996; 334: 296-301
- 14) Badesch DB et al. Ann Intern Med 2000; 132: 425-34
- 15) Olschewski H et al. N Engl J Med 2002; 347: 322-9
- 16) Simonneau G et al. Am J Respir Crit Care Med 2002; 165: 800-4
- 17) Sitbon O et al. N Engl J Med 2015; 373: 2522-33
- 18) McLaughlin VV et al. J Am Coll Cardiol 2010; 55: 1915-22
- 19) Simonneau G et al. Ann Intern Med 2008; 149: 521-30
- 20) Galiè N et al. Circulation 2009; 119: 2894-903
- 21) Tapson VF et al. Chest 2013; 144: 952-8
- 22) McLaughlin V et al. Eur Respir J 2015; 46: 405-13
- 23) Galiè N et al. N Engl J Med 2015; 373: 834-44
Standpunkte und Meinungen
- Datum des Beitrags: 31. Oktober 2017 (17:03:52)
- Verfasst von: Dr.med. Thomas Koch, Arzt und Pharmazeut ETHZ (Solothurn)
- 6 Minuten Gehstrecken-Test
Ich denke, dass in der Einleitung die Erwähnung der durchschnittlichen Normwerte für einen normalen 6 Minuten Gehstrecken-Test hätten erwähnt werden müssen. Dieser beträgt in der Durchschnittsbevölkerung mittleren Alters 700 bis 800 Meter. Es macht doch einen deutlichen Unterschied aus, wenn in einer der Studien eine Gehstreckenverlängerung von 60 Meter auf 430 Meter vorliegt, bezogen auf einen "fiktiven Normwert von 500 Meter" oder eben auf den realen Normwert von 700 bis 800 Meter
- Datum des Beitrags: 20. Oktober 2017 (19:59:10)
- Verfasst von: Dr.med. Thomas Koch, Stv.Leitender Arzt Gesundheitszentrum Solothurn (Solothurn)
- stark "under-powered" Studie (Studie 13)
Die Studie mit Epoprostenol (Flolan u.a.) ist mit 81 Personen deutlich "under-powered" ("power" knapp 50 %). In der Regel wird in Studien eine "power" von (80 % angestrebt bei einem Fehler 1.Art (Irrtumswahrscheinlichkeit, α-Fehler von 5 %, p=0,05) und einem ß-Fehler (Fehler 2.Art von 20 %). 1-ß = "power" = 80 % einer Studie. Um in dieser Studie eine "power" von 80 % zu erreichen, hätten mindestens 200 Personen einbezogen werden müssen. Diesbezüglich ist diese Studie völlig ohne Aussagekraft und irrelevant. Thomas Koch
Copyright © 2024 Infomed-Verlags-AG
PK1025
Gratisbuch bei einem Neuabo!
pharma-kritik abonnieren
-
Jahrgang 45 / 2023
Jahrgang 44 / 2022
Jahrgang 43 / 2021
Jahrgang 42 / 2020
Jahrgang 41 / 2019
Jahrgang 40 / 2018
Jahrgang 39 / 2017
Jahrgang 38 / 2016
Jahrgang 37 / 2015
Jahrgang 36 / 2014
Jahrgang 35 / 2013
Jahrgang 34 / 2012
Jahrgang 33 / 2011
Jahrgang 32 / 2010
Jahrgang 31 / 2009
Jahrgang 30 / 2008
Jahrgang 29 / 2007
Jahrgang 28 / 2006
Jahrgang 27 / 2005
Jahrgang 26 / 2004
Jahrgang 25 / 2003
Jahrgang 24 / 2002
Jahrgang 23 / 2001
Jahrgang 22 / 2000
Jahrgang 21 / 1999
Jahrgang 20 / 1998
Jahrgang 19 / 1997
Jahrgang 18 / 1996
Jahrgang 17 / 1995
Jahrgang 16 / 1994
Jahrgang 15 / 1993
Jahrgang 14 / 1992
Jahrgang 13 / 1991
Jahrgang 12 / 1990
Jahrgang 11 / 1989
Jahrgang 10 / 1988
Kennen Sie "100 wichtige Medikamente" schon?
Die Liste der 100 Medikamente sehen Sie auf der Startseite von 100 Medikamente.